Анализ геномики и биоинформатики коронавирусов

Резкое увеличение числа обнаруженных коронавирусов и секвенированных геномов коронавирусов дало беспрецедентную возможность для анализа геномики и биоинформатики этого семейства вирусов.
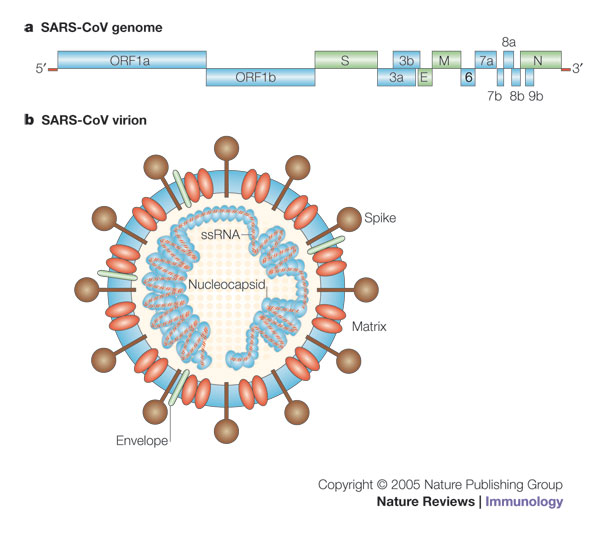
Коронавирусы обладают самыми большими геномами (от 26,4 до 31,7 т.п.н.) среди всех известных РНК-вирусов, причем содержание G + C варьируется от 32% до 43%. Различное количество небольших ORF присутствует между различными консервативными генами (ORF1ab, шип, оболочка, мембрана и нуклеокапсид) и ниже по течению от гена нуклеокапсида в различных линиях коронавируса.
Филогенетически существуют три рода: альфа-коронавирус, бета-коронавирус и гамма- коронавирус, причем бета-коронавирус состоит из подгрупп A, B, C и D. Появляется четвертый род, Deltacoronavirus, который включает коронавирус HKU11, коронавирус HKU12 и munia coronavirus HKU13.
Анализ молекулярных связей с использованием различных генных локусов показал, что время самого последнего общего предка коронавируса, связанного с SARS человека, составило еще в 1999–2002 гг., с предполагаемой частотой замещения от 4 10-4 до 2 10 -2 замен в год.
Рекомбинация в коронавирусах была наиболее заметной между различными штаммами вируса мышиного гепатита (MHV), между различными штаммами вируса инфекционного бронхита, между MHV и коронавирусом крупного рогатого скота, между кошачьим коронавирусом (FCoV) типа I и собачьим коронавирусом, генерирующим FCoV типа II, и между три генотипа человеческого коронавируса HKU1 (HCoV-HKU1).
Наблюдалось смещение использования кодонов в коронавирусах, причем HCoV-HKU1 демонстрирует наиболее экстремальное смещение, а дезаминирование цитозином и отбор клонов, подавленных CpG, являются двумя основными независимыми биологическими силами, которые формируют такое смещение использования кодонов в коронавирусах.
Традиционно вирусы были охарактеризованы и классифицированы с помощью культуры, электронной микроскопии и серологических исследований. Используя эти фенотипические методы, коронавирусы были определены как оболочечные вирусы диаметром 120–160 нм с короноподобным внешним видом. Название «коронавирус» получил из-за схожести своих пепломеров с солнечной короной. На основании их антигенных отношений коронавирусы были разделены на три группы.
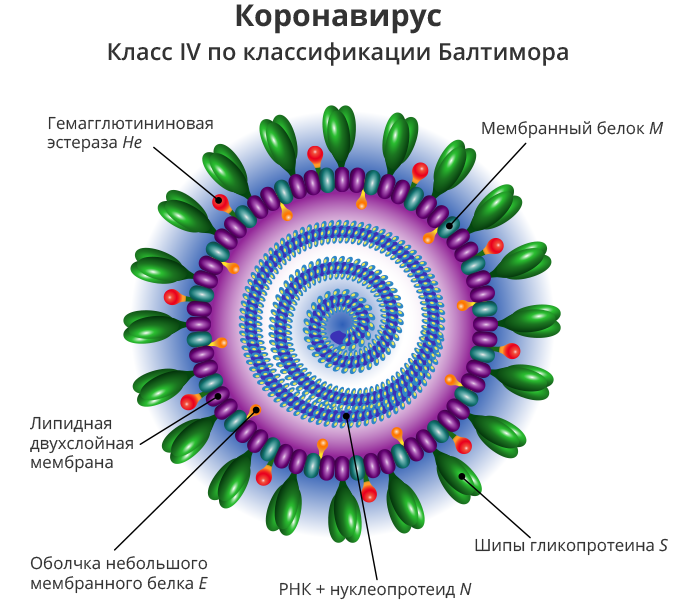
Группы 1 и 2 состоят из коронавирусов млекопитающих и птичьих коронавирусов группы 3. Изобретение и усовершенствование технологий амплификации нуклеиновых кислот, автоматизированного секвенирования ДНК и инструментов биоинформатики в последние два десятилетия произвели революцию в характеристике и классификации всех видов возбудителей инфекционных заболеваний. Используя молекулярные методы, коронавирусы классифицируются как одноцепочечные РНК-вирусы. Кроме того, результаты использования филогенетических методов для классификации также подтверждают границы групп традиционной антигенной классификации.
Филогенетические методы также позволили классифицировать связанный с SARS коронавирус (SARSr-CoV) как подгруппу группы 2, группы 2b, коронавирус; а также открытие коронавирусов группы 2c, 2d, 3b и 3c.
Недавно Исследовательская группа по коронавирусу Международного комитета по таксономии вирусов предложила три основных рода: альфа-коронавирус, бета-коронавирус и гамма-коронавирус, чтобы заменить эти три традиционные группы коронавирусов.
Первый полный геном коронавируса, вирус гепатита мыши (MHV), был секвенирован более чем через 50 лет после его выделения. До эпидемии атипичной пневмонии в 2003 году было достаточно изучены менее 10 коронавирусов с полными последовательностями генома. К ним относятся два коронавируса человека (HCoV-229E и HCoV-OC43), четыре других коронавируса млекопитающих MHV, коронавирус крупного рогатого скота (BCoV), вирус трансмиссивного гастроэнтерита (TGEV), вирус эпидемической диареи свиньи (PEDV) и один коронавирус птицы (IBV).
Эпидемия атипичной пневмонии, возникшая в южном Китае в 2003 году, повысила интерес ко всем областям исследований коронавируса, особенно к биоразнообразию и геномике коронавируса. После эпидемии атипичной пневмонии до апреля 2010 года было обнаружено 15 новых коронавирусов с секвенированием их полных геномов.
Среди этих 15 нераспознанных ранее коронавирусов были два глобально распределенных коронавируса человека: коронавирус человека NL63 (HCoV-NL63) и коронавирус человека HKU1 (HCoV-HKU1). 10 других коронавирусов млекопитающих, связанный с SARS:
- Rhinolophus bat (SARSr-Rh-BatCoV)
- Rhinolophus bat HKU2 (Rh-BatCoV HKU2)
- коронавирус Tylonycteris летучих мышей HKU4 (Ty-BatCoV HukU5 H5)
- коронавирус летучей мыши Miniopterus HKU8 (Mi-BatCoV HKU8)
- коронавирус летучей мыши Rousettus HKU9 (Ro-BatCoV HKU9)
- коронавирус Scotophilus летучей мыши 512 (Sc-BatCoV 512)
- коронавирус миниатюрной летучей мыши 1A/B (Mi-BatCo)
- коронавирус лошади (ECoV)
- коронавирус белухи SW1
- три птичьих коронавируса: коронавирус bulbul HKU11 (BuCoV HKU11), коронавирус молочницы HKU12 (ThCoV HKU12) и коронавирус мунии HKU13 (MunCoV HKU13).
Большинство из этих геномов были секвенированы с использованием РНК, извлеченной непосредственно из клинических образцов, таких как аспират или кала из носоглотки, в качестве матрицы, в то время как сами вирусы еще не культивировались.
Это обеспечило более точный анализ вирусных геномов in situ, избегая мутационного смещения во время репликации вируса in vitro. Эти последовательные усилия привели к заметному увеличению числа геномов коронавируса и дали ученым беспрецедентную возможность понять это семейство вирусов на уровне генома и in silico. Это понимание также привело к появлению новых гипотез и экспериментов в лаборатории.
В этой статье рассматривается наше современное понимание анализа геномики и биоинформатики коронавирусов. Детали инструментов биоинформатики обсуждаться не будут.
Геномика
Коронавирусы обладают самыми большими геномами: 26,4 т.п.н. (ThCoV HKU12) до 31,7 т.п.н. среди всех известных РНК-вирусов. Большой геном дал этому семейству вирусов дополнительную пластичность в приспособлении и модификации генов. Содержание G + C в геномах коронавируса варьируется от 32% (HCoV-HKU1) до 43% (Pi-BatCoV HKU5 и MunCoV HKU13). Оба 5′-и 3′-конца генома коронавируса содержат короткие нетранслируемые области. Для кодирующих областей организации генома всех коронавирусов схожесть, с характерной последовательностью генов 5′-репликазой ORF1ab, шипом (S), оболочкой (E), мембраной (M), нуклеокапсидом (N) -3′, хотя и вариабельным число дополнительных ORF присутствует в каждой подгруппе коронавируса. Мотив транскрипционной регуляторной последовательности (TRS) присутствует на 3′-конце лидерной последовательности, предшествующей большинству ORF. Считается, что мотивы TRS важны для механизма «выбора копии», который обеспечивает уникальное случайное переключение матриц во время репликации РНК, что приводит к высокой частоте гомологичной рекомбинации РНК в коронавирусах.
Филогения
Первое впечатление о филогенетическом положении штамма или вида коронавируса обычно получают путем конструирования филогенетического дерева с использованием короткого фрагмента консервативного гена, такого как Pol или N. Однако иногда это может вводить в заблуждение, поскольку результаты филогенетического анализа использования разных генов или персонажей может отличаться.
Когда SARSr-CoV был впервые обнаружен, было предложено, чтобы он составлял четвертую группу коронавируса. Однако анализ аминоконцевого домена S SARSr-CoV показал, что 19 из 20 остатков цистеина были пространственно консервативными с остатками консенсусной последовательности для бета-коронавируса.
С другой стороны, только пять остатков цистеина были пространственно законсервированы с остатками консенсусных последовательностей в альфа-коронавирусе и гамма-коронавирусе. Кроме того, при последующем филогенетическом анализе с использованием как полной последовательности генома, так и протеомных подходов был сделан вывод, что SARSr-CoV, вероятно, является ранним отщеплением от линии бета-коронавируса, аSARSr-CoV впоследствии был классифицирован как подгруппа B бета-коронавируса и историческая Бета- коронавирус как бета- коронавирусная подгруппа А.
Следовательно, филогенетическое положение коронавируса лучше всего оценивается и подтверждается путем конструирования филогенетических деревьев с использованием различных генов в геноме коронавируса. Наиболее часто используемые гены вдоль генома коронавируса для филогенетических исследований включают химотрипсин-подобную протеазу, helicase, S и N, потому что эти гены присутствуют во всех геномах коронавируса и имеют значительную длину.
Гены оболочки и мембраны, хотя и присутствуют во всех геномах коронавируса, слишком короткие для филогенетических исследований. Следует отметить, что кластер, образованный тремя новыми птичьими коронавирусами BuCoV HKU11, ThCoV HKU12 и MunCoV HKU13, который был предложен как группа 3c, может представлять новый род коронавируса, условно обозначенный как Deltacoronavirus.
Рекомбинационный анализ

В результате своего уникального случайного переключения матриц во время репликации РНК, который, как считается, опосредуется механизмом «выбора копии», коронавирусы имеют высокую частоту гомологичной рекомбинации РНК.
Впервые рекомбинация в коронавирусах была выявлена между различными штаммами MHV и впоследствии в других коронавирусах, таких как IBV, между MHV и BCoV, а также между кошачьим коронавирусом (FCoV) I типа и коронавирусом собак (CCoV). Как показано ниже, такая рекомбинация может приводить к образованию видов коронавирусов или различных генотипов внутри видов коронавирусов. По общему мнению, возможность гомологичной рекомбинации РНК и возможную часть генома, в которой произошла рекомбинация, обычно сначала оценивают с использованием бутсканированного анализа или филогенетического анализа с использованием различных частей генома коронавируса.
Другие методы для рекомбинационного анализа, такие как те в пакете RDP3, также доступны. Тогда точный тип гомологичной рекомбинации РНК лучше всего будет выявлен путем множественного выравнивания последовательностей.
Лучшим документированным примером генерации видов коронавирусов посредством гомологичной рекомбинации РНК является генерация FCoV типа II путем двойной рекомбинации между FCoV типа I и CCoV. Впервые было отмечено, что последовательность белка S в FCoV типа II была тесно связана с последовательностью CCoV, но последовательность ниже по течению от гена E в FCoV типа II была более тесно связана с последовательностью штамма FCoV типа I чем в CCoV. Это наблюдение предположило, что, возможно, произошел гомологичный случай рекомбинации РНК между геномами CCoV и FCoV типа I, что привело к генерации FCoV типа II.
Дальнейший анализ путем множественного выравнивания последовательностей выявил сайт рекомбинации в области в гене E. Несколько лет спустя была обнаружена дополнительная область рекомбинации в гене Pol, и был сделан вывод, что FCoV типа II возник в результате двух событий рекомбинации между геномами CCoV и FCoV типа I.
Что касается генерации различных генотипов у видов коронавирусов посредством гомологичной рекомбинации РНК, лучшим документально подтвержденным примером является HCoV-HKU1. Возможность гомологичной рекомбинации РНК впервые была заподозрена, когда несколько штаммов HCoV-HKU1 продемонстрировали дифференциальную кластеризацию, когда гены Pol, S и N были использованы для построения филогенетического дерева. Это наблюдение привело к последующему исследованию полного секвенирования генома 22 штаммов HCoV-HKU1.
Рекомбинационный анализ методом бутскана и филогенетический анализ с использованием различных частей 22 полных геномов выявил обширную рекомбинацию в различных частях геномов, что привело к образованию трех генотипов, A, B и C, HCoV-HKU1. Используя множественное выравнивание последовательностей, были точно определены два сайта рекомбинации.
Первый наблюдался на участке из 143 нуклеотидов около 3′-конца nsp6, где рекомбинация между генотипом B HCoV-HKU1 и генотипом C породила генотип A и второй в другом участке из 29 нуклеотидов около 3′-конца nsp16, где рекомбинация между генотипом A HCoV-HKU1 и генотипом B породила генотип C.
После эпидемии атипичной пневмонии отмечается заметное увеличение числа обнаруженных коронавирусов и секвенирование геномов коронавирусов. Это увеличение количества видов и геномов коронавирусов, всеобъемлющей и удобной для пользователя базы данных для эффективного поиска последовательностей и постоянно совершенствующихся инструментов биоинформатики позволило исследователям проводить значимый геномный, филогенетический, эволюционный уровень и расхождение, рекомбинацию и другие биоинформатические анализы на семействе Коронавирид.
Три разновидности (рода) коронавирусов: альфа-коронавирус , бета-коронавирус и гамма-коронавирус, были использованы для замены традиционных коронавирусов группы 1, 2 и 3. Вероятно, появится четвертый род, Deltacoronavirus, который включает BuCoV HKU11, ThCoV HKU12 и MunCoV HKU13. В соответствии с этой новой системой классификации коронавирусы летучих мышей доминируют над родами альфа-коронавируса и бета-коронавируса, а коронавирусы птиц доминируют над родами гамма-коронавируса и дельтакоронавируса. Это огромное разнообразие коронавирусов у летучих мышей и птиц сделало их превосходными генофондами для коронавирусов этих четырех родов.
Копирование материалов только при наличии активной ссылки на источник!
Тел.: +7(495)147-70-75 (пн-пт с 10:00 до 19:00)
Добавить комментарий
Для отправки комментария вам необходимо авторизоваться.